atomes offers to compute a number of physico-chemical properties for any 3D atomic scale models (see the list bellow).
Calculation results are handled within the software using a comprehensive graph edition system:
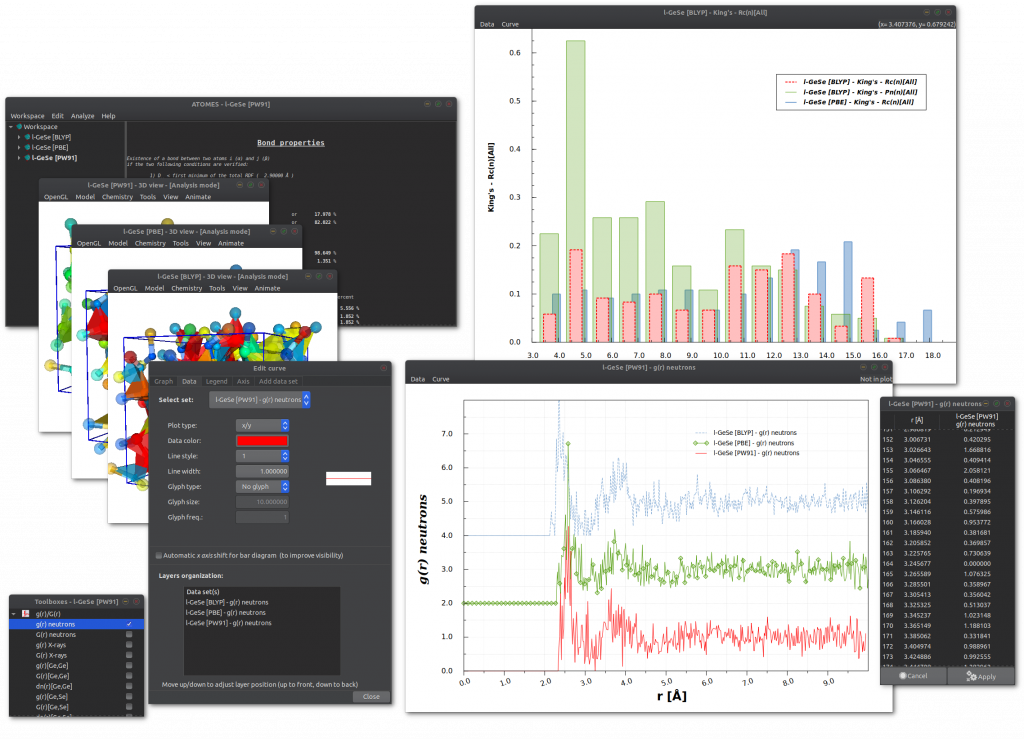
atomes regroups the following physico-chemical analyses:
- Radial distribution functions g(r) (RDFs) [1] including:
- Total RDFs for neutrons and X-rays.
- Partial RDFs.
- Bhatia-Thornton RDFs [2]
- Radial distribution functions can be computed by:
- direct real space calculation
- Fourier transforming of the structure factor calculated using the Debye formalism [3]
- Structure factors S(q) [3] including:
- Total structure factors S(q) for neutrons and X-rays.
- Total Q(q) [3], [4] for neutrons and X-rays.
- Partial S(q):
- Faber-Ziman [5] partial S(q)
- Ashcroft-Langreth [6], [7], [8] partial S(q)
- Bhatia-Thornton [9] partial S(q)
- Structure factors can be computed by:
- Fourier transforming of the radial distribution functions
- using the Debye formalism [3]
- Interatomic bond properties
- Coordination numbers
- Atomic near neighbor distribution
- Fraction of tetrahedral units
- Bond length distribution for the first coordination sphere
- Distribution of bond angles
- Distribution of dihedral angles
- Ring statistics, according to several definitions:
- All closed paths (no rules)
- King’s rings [10], [11]
- Guttman’s rings [12]
- Primitive rings [13], [14] (or Irreducible [15])
- Strong rings [13], [14]
- And including options to:
- search only for ABAB rings
- exclude rings with homopolar bonds (A-A or B-B) from the analysis
- Chain statistics, including options to:
- search only for AAAA chains
- search only for ABAB chains
- exclude chains with homopolar bonds (A-A or B-B) from the analysis
- search only for 1-(2)n-1 chains
- Spherical harmonics invariant, Ql, as local atomic ordering symmetry identifiers [7]
- Average Ql for each chemical species
- Average Ql for a user specified structural unit
- Mean Square Displacement of atoms (MSD)
- Atomic species MSD
- Directional MSD (x, y, z, xy, xz, yz)
- Drift of the center of mass
See appendix 5 of the documentation to learn more about the physics and the chemistry behind these calculations.
For more about running calculation using atomes see documentation chapter 4.
References
- M. P. Allen and D. J. Tildesley, Computer simulation of liquids. Oxford science publications, 1987.
- P. S. Salmon, J. Non-Cryst. Solids, vol. 353, pp. 2959–2974, 2007.
- M. T. Dove, M. G. Tucker, and D. A. Keen, Eur. Jour. Mat., vol. 14, pp. 331–348, 2002.
- B. Thijsse, J. App. Cryst., vol. 17, pp. 61–76, 1984.
- T. E. Faber and Z. J. M., Phil. Mag., vol. 11, no. 109, pp. 153–173, 1965.
- N. W. Ashcroft and D. C. Langreth, Phys. Rev., vol. 156, no. 3, pp. 685–692, 1967.
- N. W. Ashcroft and D. C. Langreth, Phys. Rev., vol. 159, no. 3, pp. 500–510, 1967.
- N. W. Ashcroft and D. C. Langreth, Phys. Rev., vol. 166, no. 3, p. 934, 1968.
- A. B. Bhatia and D. E. Thornton, Phys. Rev. B., vol. 2, no. 8, pp. 3004–3012, 1970.
- S. V. King, Nat., vol. 213, p. 1112, 1967.
- D. S. Franzblau, Phys. Rev. B., vol. 44, no. 10, pp. 4925–4930, 1991.
- L. Guttman, J. Non-Cryst. Solids, vol. 116, pp. 145–147, 1990.
- K. Goetzke and H. J. Klein, J. Non-Cryst. Solids, vol. 127, pp. 215–220, 1991.
- X. Yuan and A. N. Cormack, Comp. Mat. Sci., vol. 24, pp. 343–360, 2002.
- F. Wooten, Act. Cryst. A, vol. 58, no. 4, pp. 346–351, 2002.
- S. Le Roux and P. Jund, Comp. Mat. Sci., vol. 49, pp. 70–83, 2010.
- P. Steinhardt, D. R. Nelson, and M. Ronchetti, Phys. Rev. B., vol. 28, no. 2, pp. 784–805, 1983